|
|
 |
Description
|
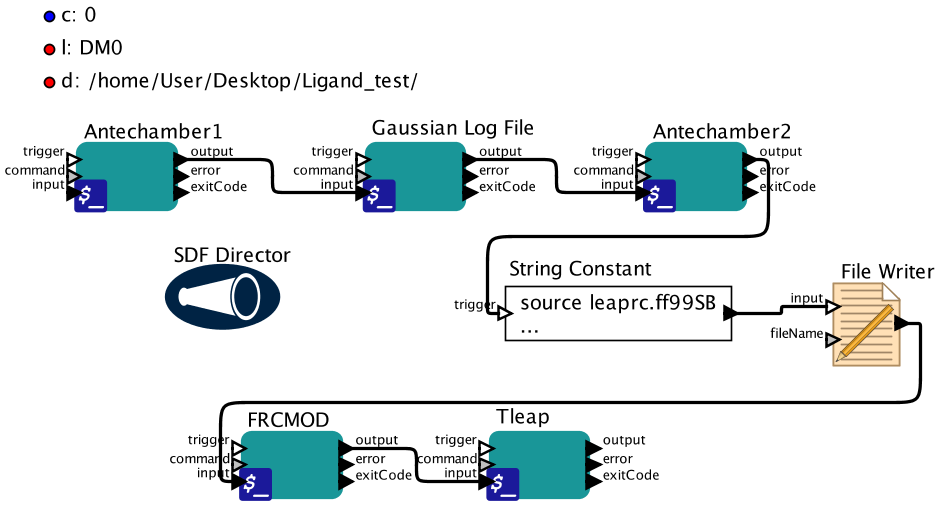
This workflow parameterizes small molecules and uses antechamber (Amber) and Gaussian09 to calculate electrostaticpotential. Amber suite and Gaussian09 must be installed in order to run this workflow. The output includesa new pdb file named LIGAND_NAME-new.pdb, prepc and frcmod files which can be used for any further experiment.
|
Required Software
|
Kepler 2.4 or above
Gaussian09
AmberTools 12 or above
|
Overview
|
None
|
Usage
|
There are two ways to run this workflow. One is through the graphic interface. After the workflow isopened, users can double-click the parameters to change their values and press the play button to start.Another way is to run Kepler via a command line. It is highly recommended that users check the valuesunder String Constant before running the workflow to ensure that the commands to execute tleap aresuitable for unique systems.
|
Parameters
|
c overall charge of the small ligand. Default is 0 which means the small molecule is neutral.
l Ligand name.
d Directory with the small molecule pdb and where all the sequential jobs will be executed.
|